
Mouse embryos showing markers of methylation
Yuelin Song/Whitehead Institute
Epigenetics: Chemical guideposts for gene expression
This story is part of our series, Looking Beyond the Gene. Click here to see all stories in this collection.
In 2012 researchers produced an extraordinary image of a single strand of DNA stretched like a tightrope between two posts, with the individual bases that compose it scarcely discernible. Although we may envision the DNA in our cells like this bare thread, the DNA within the nucleus of plant and animal cells is instead coiled around proteins, twisted into loops, and adorned with molecular tags called methyl groups. Each of these embellishments can influence whether and how genes within the DNA are expressed as proteins – the molecules integral to most cellular processes – and when and how much of those proteins are produced.
Methylation – the addition of methyl groups to DNA or DNA-associated proteins – controls gene expression by altering how the cell’s transcription machinery, which “reads” the DNA into RNA, interacts with the DNA. By affecting which genes are turned on and off, methylation helps cells define which type they are, for example, muscle, liver, or skin cells. Redistributed methylation can even change cell types as they mature. In fact, the list of methylated genes in a cell, called the methylation profile, transforms during development. Appropriate methylation is critical for health as well: Abnormal methylation can contribute to cancer, diabetes, and heart disease.
Whitehead Institute scientists are researching questions that are revealing critical aspects of methylation’s role in gene regulation, such as how methylation is maintained within a cell, how it is passed down through generations, and how a cell’s methylation profile can even be changed or repaired.
Inheriting methylation: ”She’s got your methyl groups”
Since methylation is essential for controlling gene expression and specifying particular cell types, passing down the proper placement of methylation tags from one generation of cells to the next is crucial: Too many or too few methylation tags on certain genes can have devastating consequences ranging from cell death to cancer to the inviability of a developing embryo. But little is known about how patterns of methylation marks are stably transferred – or not – from generation to generation. ROS1, a gene studied by Whitehead Institute Member Mary Gehring and Ben Williams, a postdoc in the Gehring lab, is shedding some light on this mystery. ROS1 encodes an enzyme that strips methylation from the genome of the Arabidopsis plant, and the gene’s own methylation state acts as a rheostat for proper methylation levels throughout the Arabidopsis genome.
 |
|
Read more: Epigenetic rheostat helps uncover how gene |
While methylation usually muffles a gene’s expression, in ROS1’s case methylation activates the gene and creates an important feedback loop that balances the genome’s overall methylation: When methylated, ROS1 is turned on and produces an enzyme that removes methylation from the genome, including ROS1. The lowered level of methylation switches the ROS1 gene off and halts production of the demethylating enzyme. When Williams decoupled ROS1’s methylation state from the rest of the genome, he broke the feedback loop, upsetting the plant’s methylation pattern across generations: Methylation was lost throughout the genome and progressively declined in subsequent generations, except in certain locations that were eventually remethylated. Intrigued by this delayed and location-specific remethylation, Gehring and Williams are investigating its cause, as well as other mechanisms that may be regulating this critical process.
 |
| Read more: A troubling inheritance |
As in plants, altered methylation in animal genomes can persist across generations. In order to understand if methylation might play a role in how cancer seems to run in some families, Whitehead Institute Director and Member David Page along with former postdoc Bluma Lesch discovered that mouse sperm with a mutation in a particular protein leads to modified methylation of certain portions of the sperms’ DNA and DNA-associated proteins and that these changes are linked to an increased frequency of cancer in the offspring produced from the mutated sperm. In their work, Page and Lesch deleted the Kdm6a gene from mouse sperm, whose protein removes some of the methylation from particular histone proteins around which DNA is spooled. With the demethylating KDM6A protein absent, these histone proteins became hypermethylated, and nearby portions of the sperm’s DNA are also methylated. Although most methylation in mammalian sperm and eggs is usually reset at fertilization, some of the modified sperm’s increased protein and DNA methylation were passed to the offspring, either because the methylation resisted being reset or was reestablished. The resulting mice, despite inheriting a functional Kdm6a gene from their mothers, appeared healthy until they hit middle age, but they then showed an increased likelihood of developing a wide variety of cancers, compared to mice whose methylation remained untouched. Page and Lesch have not yet investigated whether similar epigenetic inheritance occurs in humans. The question is not purely hypothetical, since certain cancer drugs currently in use target epigenetic mechanisms, and there has been no research into how altering these mechanisms could affect the children conceived by people taking the drugs.
Mother’s and father’s methylation creates tug-of-war during development
 |
|
Read more: Plant characteristics shaped by parental conflict |
In plants many seed characteristics, such as size and number of seeds produced, are both genetically and epigenetically controlled. Understanding how this happens during development could allow scientists to identify novel ways to increase crop yields. Endosperm is a plant tissue that cradles and nourishes the seed’s embryo and provides two-thirds of the calories in a typical human diet in the form of wheat grains, corn kernels, and rice grains. Endosperm development provides a window into a specific type of epigenetic gene control called imprinting, in which the copies of genes are differentially expressed based on whether they came from the seed’s mother or father. This differential expression is often associated with different DNA methylation patterns on each copy of the gene. Due to imprinting, about 200 genes have either the mother’s or father’s copy expressed in the cells of the endosperm. Imprinting is thought to have evolved because of a genetic tug-of-war between mothers and fathers. It is in the interest of the mother, whose seeds can be fertilized by numerous fathers, to nourish all of the seeds equally, no matter their father. But each father wants its seeds to outperform the seeds from other fathers and pushes the mother to devote more nutrients to his offspring. This parental tension can have significant effects. Work by Daniela Pignatta, Katherine Novitzky – former researchers in the Gehring lab – and Gehring has demonstrated that altering the imprinting of the gene HDG3 – a gene that controls the expression of other genes involved in plant patterning and development – is sufficient to affect seed size and the timing of seed development.
Disease in a Petri dish: Resetting methylation to create disease models using induced pluripotent stem cells
 |
|
Read more: Reprogrammed fibroblasts identical to |
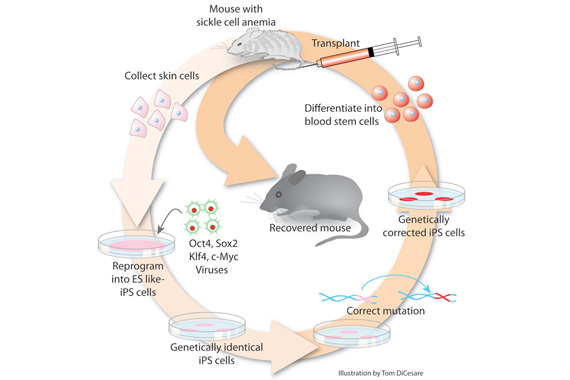
In plants and animals, methylation changes are one aspect that define an embryo’s development. Scientists largely took for granted that this developmental path was a one-way street until 2006, when, using a cocktail of four “reprogramming factors”, the first mouse skin cells were pushed back to an embryonic stem cell-like state. These “induced pluripotent stem cells” (iPSCs) have the plasticity to develop into virtually every cell type, including those that are difficult to study due to their inaccessibility and limited numbers, such as the neurons that wither and die deep in the brains of Parkinson’s disease patients. Curious as to whether iPSCs could be used as a tool to model disease in the lab, Whitehead Institute Founding Member Rudolf Jaenisch pivoted a portion of his lab’s focus into studying this new type of cell. Soon after, Jaenisch and a team of scientists, including former Jaenisch lab postdoctoral researchers Marius Wernig, Alexander Meissner, and Tobias Brambrink; former graduate student Ruth Foreman; and Manching Ku, a research fellow from Bradley Bernstein's lab at Massachusetts General Hospital, significantly improved on the initial approach and created iPSCs that are functionally equivalent to mouse embryonic stem cells.
 |
|
Read more: Reprogrammed adult cells treat sickle-cell |
According to Jaenisch, successful reprogramming requires resetting a cell’s genome-wide methylation profile to that of a pluripotent stem cell. And by using specific proteins, scientists can coax iPSCs – and their methylation patterns – to become almost any other cell type, such as muscle, bone, or nerve cells. This flexibility could one day enable scientists to create pancreatic cells to replace those impaired by diabetes, regenerate nerves, and grow replacement organs from a patient’s own cells. But for Jaenisch, iPSCs currently hold the most power for modeling patient-specific diseases in animals or the Petri dish. By taking a small skin tissue sample from a person with Parkinson’s disease, reprogramming those cells to an iPSC state, and prodding them to develop into neurons, Jaenisch and Frank Soldner and Dirk Hockemeyer, former postdocs in the Jaenisch lab, created the type of neural cells that are affected in Parkinson’s. Using iPSCs, scientists can create sufficient numbers of those cells that can be studied in the lab, allowing scientists to research the mechanisms involved in the disease. Jaenisch has used iPSCs to model other diseases, such as sickle cell anemia, and the rare genetic disease Niemann-Pick type C (NPC), and says that iPSCs are a potent tool for scientists investigating blood disorders and autoimmune diseases, such as multiple sclerosis and type 1 diabetes as well.
Correcting disease-causing methylation
Methylation influences gene expression and cell state, and abnormal methylation patterns have been linked to many diseases, such as neurological disorders and cardiovascular disease. Altering the methylation on a particular gene might prevent or stop such a disorder, but such surgical changes have been difficult because scientists had no tool to flip methylation states without affecting other parts of the genome. By adapting the CRISPR/Cas9 genome editing system, Jaenisch lab postdocs Shawn Liu and Hao Wu are able to precisely add or remove methylation from specific sites on the genome in living mouse neural cells and leave the rest of the genome untouched.
 |
|
Read more: Fragile X syndrome neurons restored using |
Liu and Jaenisch then used this approach to tackle the Fragile X syndrome disease gene, FMR1. Affecting 1 out of 3600 boys born, Fragile X is the most frequent cause of intellectual disability in males. The disorder is caused by excessive methylation of FMR1, which shuts off its function. In neurons derived from Fragile X syndrome iPSCs, Liu removed the excess methylation, which revived the FMR1 gene’s expression to normal levels and reversed the abnormal electrical activity in neurons that is associated with the syndrome. When Liu engrafted rescued neurons into the brains of mice, the FMR1 gene remained active in the neurons for at least three months, suggesting that the corrected methylation may be sustainable in an animal. This technique’s success in restoring function to Fragile X neurons indicates it may also prove useful for other diseases caused by abnormal methylation, such as facioscapulohumeral muscular dystrophy and imprinting diseases.
A methyl group is comprised of four atoms, but as a component of DNA methylation, it is a powerful regulator of gene expression. By studying and molding methylation states, Whitehead Institute scientists are at the forefront of research to understand the effects of methylation and the mechanisms that control it. As Gehring, Page, and Jaenisch investigate methylation’s role in the cell, their work is inspiring novel ways that methylation could be used as a tool to decipher previously unknown aspects of gene regulation and positively impact health.
Contact
Communications and Public Affairs
Phone: 617-452-4630
Email: newsroom@wi.mit.edu


